INHIBITOR MECHANISM OF ACTION
Saquinavir was optimized to be a potent competitive inhibitor of HIV-1 protease, but that does not mean the drug is without limitations, particularly when it comes to rapidly evolving viral resistance.
Human immunodeficiency virus (HIV) is a virus that when not under control has the potential to take over a person’s immune system and removes its ability to fight even simple diseases. HIV itself, however, lacks proofreading mechanisms and is thus more susceptible to mutation than would take place in the genome of a living system.(1) Some mutations are detrimental to the survival of the virus, and these residues are highly conserved. But every other non-essential residues in HIV proteins are vulnerable to mutations that lead to a variety of amino acid polymorphisms. The presence of all of these HIV variants make HIV a difficult entity to drug. Saquinavir is a drug that has been effective in the treatment of HIV. It is a potent protease inhibitor, and one of the first effective ones at that. However, this drug often leads to common mutations that easily render it ineffective.(2)
METHOD OF ADMINISTRATION
In regards to delivery of saquinavir to the human body, the current FDA approved use of saquinavir in the market drugs Fortovase and Invirase utilizes the active compound saquinavir mesylate (Figure 1), which is the salt of the free, active base saquinavir.(3) As is common with many first-generation protease inhibitors, the bioavailability of orally administered saquinavir is about 4%.(3) To combat this incomplete absorption, saquinavir is required to be co-administered with ritonavir, which effectively acts to inhibit the enzymes involved in saquinavir's first-pass metabolism.(3) As such, saquinavir is only currently approved for administration as a free base and not a prodrug, indicating that it is possible for the compound to be active unmodified, but its bioavailability is extremely low. In the 25 years since the drug’s approval, there have been many studies into potential prodrug modifications of saquinavir that result in greater absorption and higher bioavailability. Specific studies have included the use of l-Leucine and l-phenylalanine conjugates,(4) D-glucose- and PEG-conjugates,(5) and a vitamin C derivative, ascorbyl-succinic-saquinavir, which targets sodium dependent vitamin C transporter (SVCT).(6) As the development of new, more effective HIV drugs continues, it will be interesting to see if these chemical biology studies into prodrugs of first-generation protease inhibitors will result in any new FDA-approved therapeutics, or if saquinavir will be figuratively left on the shelf, unmodified since 1995.
Figure 1. The structure of saquinavir mesylate, the salt of the free, active base and the compound currently approved for use by the FDA. Although saquinavir is not currently administered as a prodrug, there have been numerous research studies into forming prodrugs to increase bioavailability of the compound when orally administered.

SAQUINAVIR STRUCTURE - THE KEY TO FUNCTION
SAQUINAVIR IS A TRANSITION STATE ANALOGUE
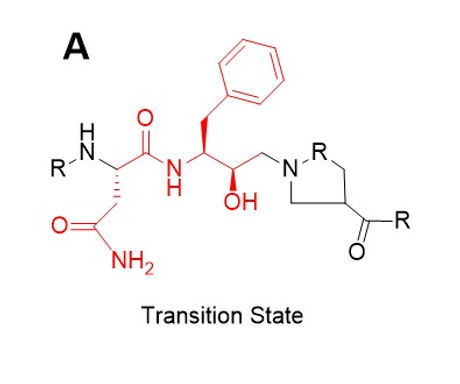
One driver of saquinavir’s inability to be effective in the presence of specific mutations is because of its intentional design. Saquinavir was designed using the transition state of HIV protease: a hydroxyethylamine.(7) This intermediate forms as a result of the reaction to cleave the Proline-Phenylalanine amide bond of a protein.(7) The non-essential moieties of the transition state were then adjusted to maximize selectivity and potency, the comparison of which can be seen in Figure 2.
COMPARE THE TRANSITION STATE OF THE NORMAL SUBSTRATE AND THE STRUCTURE OF SAQUNIAVIR
Figure 2. Naturally occurring transition state (A) and saquinavir (B), a transition state analog that inhibits protease activity. The unchanged part of the structure, the transition state analog portion, is highlighted in red. This part of the molecule consists of the Phe-Pro bond that is ultimately cleaved.

MECHANISM OF COMPETITIVE INHIBITION
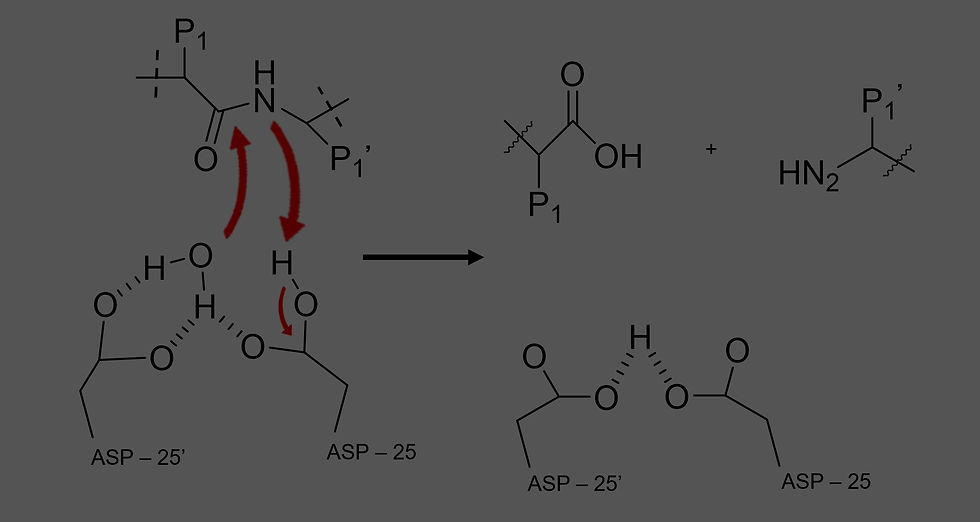
As detailed above, saquinavir is a peptidomimetic inhibitor developed from the hydroxyethylamine transition state of the hydrolysis reaction between two hydrophobic amino acids of a polypeptide.(8) A transition state analogue, saquinavir acts as a competitive inhibitor that works to outcompete the Gag and Gag-Pol polyprotein substrates which normally undergo proteolysis via catalysis by HIV-1 protease.(9) As such, the binding of the target enzyme and the antiviral agent is dependent on the very residues of HIV-1 protease most important in its catalytic activity as an aspartic acid protease: the two aspartic acid residues (Asp – 25 and Asp – 25’) that form the catalytic dyad at the interface of the homodimer.(9) As seen in the generic mechanism of normal HIV-1 protease function (Figure 3), the P1 and P1’ side chains of the usual polypeptide substrate can instead be replaced by the moieties surrounding the amide bond of saquinavir, as can be seen color coded in Figure 4. Specifically, X-ray crystallography (see Figure 5) has shown that the methylene groups of this inhibitor fit into the active site much differently than the normal protein substrate, with the carbonyl of the [(4aS,8aS)-decahydroisoquinolin-3(S)-yl]carbonyloxy (DIQ) group binding to the water molecule and connecting the inhibitor with the flap regions and body of the enzyme.(10) In short, Saquinavir mimics the scaffold of the normal peptide bond in the substrate, but it itself cannot be cleaved, effectively outcompeting the normal substrate such that HIV-1 protease cannot hydrolyze the Gag and Gag-Pol polyproteins and immature, non-infectious viral particles are produced.(3)

Figure 3. The P1 and P1’ hydrophobic side chains of the polypeptide substrate can be made general by replacement with the moieties surrounding the peptide bond of saquinavir. Saquinavir will complex with the HIV-1 protease active site but not undergo hydrolysis as seen in this general mechanism, thereby acting as a competitive inhibitor.
OPTIMIZATION OF STRUCTURE
During optimization, the terminal groups of the molecule were changed to achieve the greatest combination of potency and selectivity. Beginning with selectivity, the drug is naturally selective to only HIV proteases because proline amide bonds are not cleavable by mammalian endopeptidases.(11) Thus, this structure is not recognizable as a target by native proteases to the human body. The initial target for optimization included the phenylalanine residue adjacent to a reduced amide, but the presence of the reduced amide proved to be ineffective for use as an inhibitor.(11) The hydroxyethlyamine group was efficiently potent and selective, but just that group with the phenylalanine residue was too small to achieve sufficient inhibition. Slight elongation with a proline –like group and one more amide bond on the other side gave the drug in progress ample mass for effective inhibition.(11) In addition, the binding pocket of HIV protease contains a large hydrophobic pocket that the bulky aromatic moiety in saquinavir binds to.(11) This hydrophobic region of the drug likely allows for more effective binding of the inhibitor to the active site. Last, the slight variant of proline with the nonaromatic ring structure and tert-butyl group increase potency of the inhibitor without introducing inhibition to human endopeptidases.(11) To sum this up, saquinavir was designed using the known transition state and substrate of HIV proteases. Several modifications taking advantage of certain aspects of the protease structure such as the hydrophobic pocket and other group introduction have yielded an incredibly potent protease inhibitor that has been effectively treating HIV for years.
VIRAL RESISTANCE TO SAQUINAVIR RESULTS IN A DECREASE IN INHIBITION EFFICACY
Saquinavir is still in use today because of it relatively benign side effects compared to other drug regiments, but its biggest flaw is most certainly its inability to adapt to specific mutated proteases.(12) HIV regularly becomes resistant to saquinavir, and that resistance can be attributed to two distinct saquinavir mutations: G48V and/or L90M.(12) The crystal structure of saquinavir, particularly those with bound saquinavir, have been particularly telling as to why these mutations are the most detrimental to the effectiveness of saquinavir, one of those structures is seen in Figure 6.
Within the structure, G48 is located within the active site of HIV protease and interacts with the flap that effectively can act as a door to strengthen the interaction between bound saquinavir and HIV protease while L90 is not located within the active site but appears to have significant structural roles for the enzyme, both of which can be observed in blue in Figure 6.(13) When either a G48V mutation and/or a L90M mutation occurs, the binding affinity of saquinavir drops significantly (13.5 fold for a G48V mutation and 41-fold decrease for the double mutation).(13) This drop makes it nearly impossible for saquinavir to bind to the protease, which renders it ineffective.
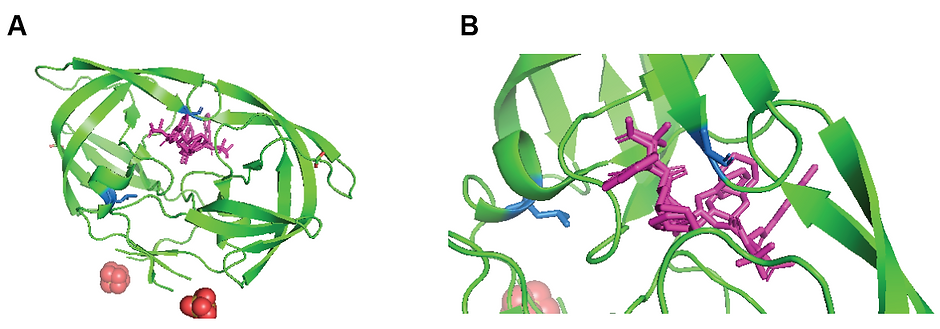
Figure 6. Crystal structure of saquinavir bound HIV protease. (A) Full protein view where the dimerization of the protease is clearly seen with the inhibitor in purple bound between monomers. (B) Zoomed in view of the active site bound to saquinavir (purple). G48 and L90, two primary residues that when mutated are responsible for saquinavir resistant HIV are colored in blue. PDB: 3OXC
The decreased ability for saquinavir to bind to HIV protease is driven by the structural changes of the protease. The G48V mutation decreases the flexibility of the protease’s flap, which prevents the flap from trapping saquinavir in the active site, and even if the inhibitor manages to bind, the larger valine side chain wedges itself between the quinolone ring structure (hydrophobic substituent) and the phenylalanine residue which alters the binding orientation and decreases bond strength.(2,13) An L90M mutation however makes binding of saquinavir even more difficult because it subtly alters the entire structure of the protease. Consequentially, the active site is more difficult to access and the catalytic residue, D25, is no longer in an optimal orientation to bound saquinavir even if the drug manages to bind.(2,13) In combination, the double mutant results in a smaller binding pocket that makes it extremely difficult for the inhibitor to bind, it stabilizes the protease conformation to one that is in between those of unbound HIV protease and bound HIV protease, which eliminates the protein’s ability to “snap” into a bound conformation to allow for catalysis.(2)
Some things to note about saquinavir and its two main mutations leading to saquinavir resistance are that first, HIV is bound to mutate. From a scientific perspective, it is more a question of when and where the mutation will take place. With that being said, analysis of the most common amino acid polymorphisms of HIV protease reveal that the G48V and L90M mutations appear significantly more often in saquinavir treated HIV protease than in drug naïve HIV proteases.(1) This means that these specific mutations can be considered drug driven mutations because they confer a survival advantage to HIV in the presence of this specific drug. Another interesting part of the saquinavir resistance story is that the rise of these mutations are different when studied in vivo vs when it is studied in vitro. When studied in vitro, the G48V mutation almost always arises first with the L90M mutation to follow, the double mutant only rarely occuring.(12) In vivo mutations for these specific residues occurs in the opposite manner, however, with the L90M mutation always observed first.(12) Last, because saquinavir is a transition state analog, it cannot adapt itself to these mutations as it was designed to be most effective in the active site of wild type HIV-protease.(1) But, saquinavir still proves to be incredibly potent and effective up until the development of these mutations. Thus, saquinavir still remains a key drug in the treatment of HIV, but current development of new protease inhibitors has hopes to identify an inhibitor that targets the conserved residues essential to the survival of HIV.(1) This way, HIV is free to mutate but the protease inhibitor will still remain effective.
ALL IN ALL, SAQUINAVIR WAS REVOLUTIONARY IN THE TREATMENT OF HIV AND CONTINUES TO BE A STAPLE IN HIV TREATMENT DESPITE ITS PROPENSITY FOR MUTATIONS THAT CAUSE IT TO BE INEFFECTIVE. FORTUNATELY, HIV TREATMENT HAS PROGRESSED SO THAT WHEN RESISTANCE ARISES TO ONE DRUG, IT IS POSSIBLE TO INTRODUCE A NEW INHIBITOR THAT TARGETS THE PROTEASE IN A DIFFERENT MANNER, AND TREATMENT WITH PROTEASE INHIBITORS CAN CONTINUE TO BE SUCCESSFUL.
REFERENCES
Ohtaka, H. Thermodynamic Rules for the Design of High Affinity HIV-1 Protease Inhibitors with Adaptability to Mutations and High Selectivity towards Unwanted Targets. Int. J. Biochem. Cell Biol. 2004, 36 (9), 1787–1799. https://doi.org/10.1016/j.biocel.2004.02.021.
Hong, L.; Zhang, X. C.; Hartsuck, J. A.; Tang, J. Crystal Structure of an in Vivo HIV-1 Protease Mutant in Complex with Saquinavir: Insights into the Mechanisms of Drug Resistance. Protein Sci. 2000, 9 (10), 1898–1904. https://doi.org/10.1110/ps.9.10.1898.
Saquinavir https://go.drugbank.com/drugs/DB01232 (accessed Nov 6, 2020).
Gaucher, B.; Rouquayrol, M.; Roche, D.; Greiner, J.; Aubertin, A.-M.; Vierling, P. Prodrugs of HIV Protease Inhibitors-Saquinavir, Indinavir and Nelfinavir-Derived from Diglycerides or Amino Acids: Synthesis, Stability and Anti-HIV Activity. Org Biomol Chem 2004, 2 (3), 345–357. https://doi.org/10.1039/b313119j.
Rouquayrol, M.; Gaucher, B.; Roche, D.; Greiner, J.; Vierling, P. Transepithelial Transport of Prodrugs of the HIV Protease Inhibitors Saquinavir, Indinavir, and Nelfinavir Across Caco-2 Cell Monolayers. Pharm Res 2002, 19 (11), 1704–1712. https://doi.org/10.1023/A:1020913631309.
Luo, S.; Wang, Z.; Patel, M.; Khurana, V.; Zhu, X.; Pal, D.; Mitra, Ashim. K. Targeting SVCT for Enhanced Drug Absorption: Synthesis and in Vitro Evaluation of a Novel Vitamin C Conjugated Prodrug of Saquinavir. Int J Pharm 2011, 414 (0), 77–85. https://doi.org/10.1016/j.ijpharm.2011.05.001.
Moyle, G. Drug Evaluation: Anti-Infectives: Saquinavir: A Review of Its Development, Pharmacological Properties and Clinical Use. Expert Opin. Investig. Drugs 1996, 5 (2), 155–167. https://doi.org/10.1517/13543784.5.2.155.
National Center for Biotechnology Information. PubChem Compound Summary for CID 441243, Saquinavir https://pubchem.ncbi.nlm.nih.gov/compound/441243 (accessed Nov 6, 2020).
Ghosh, A. K.; Chapsal, B. D. Chapter 13 - Design of the Anti-HIV Protease Inhibitor Darunavir. In Introduction to Biological and Small Molecule Drug Research and Development; Ganellin, R., Roberts, S., Jefferis, R., Eds.; Elsevier: Oxford, 2013; pp 355–384. https://doi.org/10.1016/B978-0-12-397176-0.00013-3.
Krohn, A.; Redshaw, S.; Ritchie, J. C.; Graves, B. J.; Hatada, M. H. Novel Binding Mode of Highly Potent HIV-Proteinase Inhibitors Incorporating the (R)-Hydroxyethylamine Isostere. J. Med. Chem. 1991, 34 (11), 3340–3342. https://doi.org/10.1021/jm00115a028.
Roberts, N. A.; Martin, J. A.; Kinchington, D.; Broadhurst, A. V.; Craig, J. C.; Duncan, I. B.; Galpin, S. A.; Handa, B. K.; Kay, J.; Kröhn, A.; Lambert, R. W.; Merrett, J. H.; Mills, J. S.; Parkes, K. E. B.; Redshaw, S.; Ritchie, A. J.; Taylor, D. L.; Thomas, G. J.; Machin, P. J. Rational Design of Peptide-Based HIV Proteinase Inhibitors. Science 1990, 248 (4953), 358–361.
Jacobsen, H.; Hanggi, M.; Ott, M.; Duncan, I. B.; Owen, S.; Andreoni, M.; Vella, S.; Mous, J. In Vivo Resistance to a Human Immunodeficiency Virus Type 1 Proteinase Inhibitor: Mutations, Kinetics, and Frequencies. J. Infect. Dis. 1996, 173 (6), 1379–1387. https://doi.org/10.1093/infdis/173.6.1379.
Saen-oon, S.; Aruksakunwong, O.; Wittayanarakul, K.; Sompornpisut, P.; Hannongbua, S. Insight into Analysis of Interactions of Saquinavir with HIV-1 Protease in Comparison between the Wild-Type and G48V and G48V/L90M Mutants Based on QM and QM/MM Calculations. J. Mol. Graph. Model. 2007, 26 (4), 720–727. https://doi.org/10.1016/j.jmgm.2007.04.009.